






Investigadores diseñan un modelo informático en 3-D del genoma humano
La secuenciación del genoma es un hito en la biología moderna, ya que permite el acceso a la "lista de instrucciones" completo
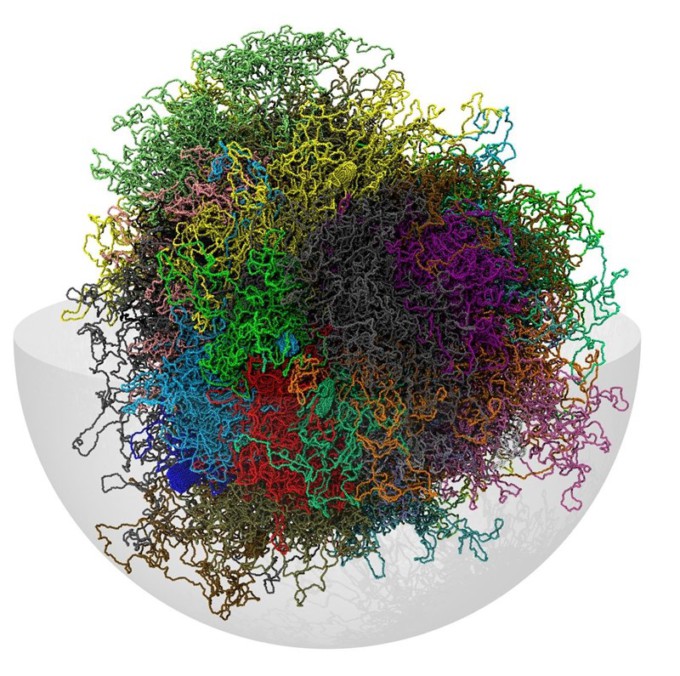
Un grupo coordinado por la Escuela Internacional de Estudios Avanzados (SISSA, por sus siglas en inglés) de Trieste, en Italia, ha construido un modelo informático tridimensional del genoma humano. La forma del ADN (así como su secuencia) afecta significativamente a los procesos biológicos y, por tanto, es crucial para entender su función.
Este nuevo estudio ha proporcionado un primer retrato robot tridimensional aproximado pero realista del genoma humano. Gracias a las características del nuevo método, la reconstrucción estructural basada en la información experimental y métodos estadísticos se perfeccionará a medida que se disponga de nuevos datos experimentales.
Los detalles de este trabajo, que fue realizado en colaboración con expertos de la Universidad de Oslo, en Noruega, se revelan en un artículo publicado esta semana en 'Scientific Reports', una publicación perteneciente al grupo editorial de 'Nature'.
La secuenciación del genoma es un hito en la biología moderna, ya que permite el acceso a la "lista de instrucciones" completo (la secuencia química de la composición genética) para el desarrollo y la función de los organismos. La secuenciación del genoma es un poco como escribir el orden exacto del color de las cuentas de un collar: saber cómo se organizan a lo largo del hilo no nos da ninguna indicación sobre a la forma del collar.
La forma de la cadena de ADN puede ser muy compleja, dado que los cromosomas se disponen con holgura en una maraña aparentemente caótica en el núcleo celular. Como la forma de los cromosomas puede tener un efecto decisivo en su función, es importante caracterizarla, en parte porque los científicos creen que la maraña de ADN en el núcleo es sólo aparentemente caótica y que tiene lugar una "geografía" específica para cada tejido y etapa de la vida celular.
"Llegar a una descripción precisa de la forma de la maraña de ADN es por desgracia muy complicado --explica Cristian Micheletti, profesor en SISSA y coordinador del nuevo estudio--. En nuestro caso, hemos utilizado datos experimentales sobre 'parejas de proximidad".
"Imagínese tener que crear un mapa de una ciudad -explica-- basándose únicamente en información como que 'la oficina de correos se encuentra frente a la estación', 'el farmacéutico está cerca del gimnasio', 'el mercado de frutas y verduras se encuentra cerca del campo de fútbol ' y así sucesivamente. Si usted tiene sólo un pequeño número de estas explicaicones, el mapa será aproximado y, en algunos casos, indeterminado. Pero si usted tiene cientos, miles o incluso más, entonces su mapa será cada vez más preciso y exacto. Ésta es la lógica que seguimos".
GRACIAS A LA INFORMACIÓN 'DE LOS VECINOS' EN EL ADN
"Las parejas de proximidad', por tanto, se refiere a la información sobre la cercanía de dos puntos en el mapa. En el caso del ADN nuclear, esta información fue proporcionada por una técnica (que Micheletti define como "brillante"), conocida como Hi-C, desarrollado por equipos de investigación de América del Norte en 2010. En esta técnica físico-química, partes del genoma situadas cerca unas de otras en el núcleo se unen y entonces se identifican por su secuencia", relata este experto.
"Mediante la recopilación de un gran número de estos pares de proximidad, los científicos descubrieron qué puntos de los cromosomas se encuentran cerca unos de otros en el núcleo. Aunque es la técnica actual más poderosa para investigar la organización del ADN en el núcleo, todavía es insuficiente para inferir su forma general. Por esta razón, pensamos que habría que tratar de ir más allá", comenta Micheletti.
"Se utilizó una base de datos pública de pares de proximidad inicialmente derivados de un solo experimento Hi-C. La base de datos contiene información sobre cientos de miles de pares de proximidad", detalla Marco Di Stefano, investigador que completó su doctorado en SISSA en 2014 (con este proyecto), primer autor del artículo y actualmente investigador postdoctoral en el Centro Nacional de Análisis Genómica de Barcelona. Los autores crearon un modelo virtual de grano grueso de todos los cromosomas en una conformación tridimensional "básica" e identificaron la posición de los dos fragmentos de ADN de cada par de proximidad para dibujarlos el uno junto al otro de manera apropiada por la curva de la hebra.
"Al repetir esta operación para todos los pares de proximidad conocidos experimentalmente se obtuvo una estructura enmarañada, aunque no al azar, que reveló la forma de todos los cromosomas del genoma humano, que estaba oculta dentro de los datos --explica Di Stefano--. No hace falta decir que cuanto mayor es el número de pares que se utilizan, más preciso es el modelo 3D resultante".
De hecho, después de esta primera fase de Micheletti y sus colegas añadieron una nueva serie de datos experimentales al modelo. "Justo cuando estábamos trabajando en el proyecto, se publicó un nuevo conjunto más detallado de datos de Hi-C, por lo que hemos utilizado esos también", agrega Micheletti.
"A decir verdad, tuvimos algunas preocupaciones de que nuestro nuevo método pudiera no ser lo suficientemente robusto y que el nuevo conjunto de datos podría entrar en conflicto con el modelo 3D y arruinar lo conseguido previamente. Sin embargo, casi para nuestra sorpresa, vimos que la conformación permaneció bastante similar", relata.
"Los nuevos datos simplemente refinaron el modelo y casi por arte de magia las distintas zonas de los cromosomas se posicionaron en los lugares correctos del núcleo. Esto fue aún más convincente después de haber tenido éxito en la descripción de los datos reales con un buen grado de aproximación y esperamos que en el futuro los datos recogidos nos permitan revelar con detalle creciente la forma en la que el ADN está encerrado en nuestras células", augura.


